Rate Models (Reading:
Faraway (2006), section 3.2)
Purott and Reeder (1976) presented some
data from an experiment conducted to
determine the effect of gamma radiation on the number of chromosomal
abnormalities (ca)
observed. The number of cells exposed in each run (cells,
in hundreds) differs. The dose amount (doseamt)
and the rate (doserate)
at which the dose is applied are the predictors of interest. Let us read the
data into R and take a look at it:
> dicentric <- read.table("dicentric.txt")
> dicentric
cells ca doseamt
doserate
1 478 25 1.0
0.10
2 1907 102 1.0
0.25
3 2258 149 1.0
0.50
…deleted…
27 144 206 5.0
4.00
Notice that:
-
we can model the response as binomially distributed,
i.e., yx~binomial(100nx,
px),
where yx
is ca and
nx
is cells, and use
appropriate link function and Xb
structure to relate px
to the predictors. In other words, we can apply a binomial GLM to the data.
-
because the values of yx/(100nx)
are small (its maximum value is
1.43/100=0.0143) as shown in the table below and
the values of 100nx
are relatively large, a Poisson approximation to the binomial is effective,
i.e., a Poisson GLM is an effective alternative for the data
-
however, because the values of
nx
differs, it does not make much sense to build a Poisson GLM using
ca as the response and
doseamt and
doserate as the
predictors (Q: how to interpret the value of
prediction if we fit such a model?)
-
therefore, nx,
which is the size variable in this case, should play a role in the Poisson GLM so that meaningful conclusions can be drawn
Let us take a look at how the proportions,
yx/nx,
change with the two predictors:
>
round(xtabs(ca/cells ~ doseamt+doserate, dicentric),2)
doserate
doseamt 0.1 0.25
0.5 1 1.5 2 2.5 3 4
1 0.05 0.05 0.07
0.07 0.06 0.07 0.07 0.07 0.07
2.5 0.16 0.28 0.29
0.32 0.38 0.41 0.41 0.37 0.44
5 0.48 0.82 0.90
0.88 1.23 1.32 1.34 1.24 1.43
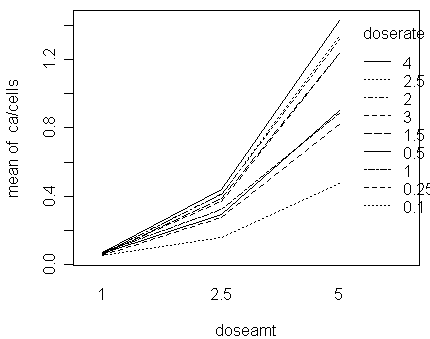
We can see that:
-
the proportion increases with
doserate and
doseamt
-
the effect of doserate
seems larger than doseamt
-
furthermore, the effect of
doserate is not linear
-
its influence could be multiplicative, i.e.,
doserate
influences ca/cells
through multiplication rather than addition
-
a log transformation can sometimes
simplify a multiplicative model
-
exercise: draw
some scatter plots to check whether the relationship between
ca/cells
and log(doserate)
is more linear
-
we will replace
doserate by
log(doserate) in the
following analysis
-
it is clear that some interaction effects are significant
-
however, if we replace ca/cells
by log(ca/cells),
the interactions can possibly be reduced (or even removed in some cases) (Q:
can you see why?)
-
because log[yx/(100nx)]»logit[yx/(100nx)] when yx/(100nx)
is small, we can expect that the result of fitting a Normal linear model to
log(ca/cells)
would be similar to that of fitting a binomial GLM with logit link to
ca
-
exercise: use a Normal linear
model to relate the response
log(ca/cells)
to the predictors and give your opinions on its pro's and con's
-
an interaction plot can offer a good visual summary of
the 4 points given above:
>
with(dicentric,interaction.plot(doseamt,doserate,ca/cells))
Let us treat
log(doserate) as a continuous
variable and dosamt as
a categorical variable and fit a Normal linear model to the response
ca/cells:
-
Q: why
dosamt treated as
categorical?
-
notice that dosamt
has 3 levels and its effect on
ca/cells cannot be explained only by a linear
effect as shown in the interaction plot. Therefore, no matter we use the
linear-quadratic coding or the dummy-variable coding, the two effects coded
from dosamt would
be both significant and span the same column space
-
on the other hand,
log(doserate) has 9 levels but its effect on
ca/cells seems
only linear. If we use the dummy-variable coding, it will require 7 more
degrees of freedom to explain the difference between levels
>
dicentric$dosef <- factor(dicentric$doseamt)
>
lmod <- lm(ca/cells ~ log(doserate)*dosef, dicentric)
>
summary(lmod)
Call:
lm(formula = ca/cells ~
log(doserate) * dosef, data = dicentric)
Residuals:
Min 1Q
Median 3Q Max
-0.184275 -0.004212
0.001314 0.021208 0.089076
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept)
0.063489 0.019528 3.251 0.00382 **
log(doserate)
0.004573 0.016692 0.274 0.78680
dosef2.5
0.276315 0.027616 10.005 1.92e-09 ***
dosef5
1.004119 0.027616 36.359 < 2e-16 ***
log(doserate):dosef2.5
0.063933 0.023606 2.708 0.01317 *
log(doserate):dosef5
0.239129 0.023606 10.130 1.54e-09 ***
---
Signif. codes: 0 '***'
0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error:
0.05858 on 21 degrees of freedom
Multiple R-Squared:
0.9874, Adjusted R-squared: 0.9844
F-statistic: 330 on 5
and 21 DF, p-value: < 2.2e-16
As can be seen from the R2,
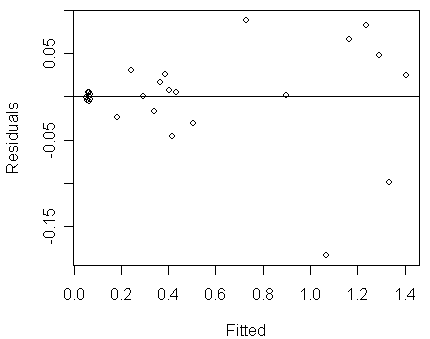
the model fits well. However, a look at the diagnostics reveals a problem:
>
plot(residuals(lmod) ~ fitted(lmod),xlab="Fitted",ylab="Residuals")
>
abline(h=0)
Notice that:
-
we can see clear evidence of non-constant variance (Var(yx)
increases with E(yx)).
-
we might prefer an approach that directly model the count
response, say ca~Poisson.
However, the size variable nx
should be appropriately included in the model so that its effect can be
correctly adjusted for
Let us consider the Poisson model, yx~Poisson(mx),
with the link function:
log(mx/nx)=hx=Xb
Û
log(mx)=log(nx)+Xbºhx'.
The log(nx)
is now regarded as a predictor with the coefficient fixed as one. In this
manner, we are modeling the rate of chromosomal abnormalities while still
maintaining the count response for the Poisson model. In R, we can fix the
coefficient as one by using an offset,
which is a term on the predictor side with no parameter attached:
>
rmod <- glm(ca ~ offset(log(cells))+log(doserate)*dosef, family=poisson,dicentric)
>
summary(rmod)
Call:
glm(formula = ca ~
offset(log(cells)) + log(doserate) * dosef, family = poisson, data =
dicentric)
Deviance Residuals:
Min 1Q
Median 3Q Max
-1.49101 -0.62473
-0.05078 0.76786 1.59115
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept)
-2.74671 0.03426 -80.165 < 2e-16 ***
log(doserate)
0.07178 0.03518 2.041 0.041299 *
dosef2.5
1.62542 0.04946 32.863 < 2e-16 ***
dosef5
2.76109 0.04349 63.491 < 2e-16 ***
log(doserate):dosef2.5
0.16122 0.04830 3.338 0.000844 ***
log(doserate):dosef5
0.19350 0.04243 4.561 5.1e-06 ***
---
Signif. codes: 0 '***'
0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter
for poisson family taken to be 1)
Null deviance:
4753.00 on 26 degrees of freedom
Residual deviance:
21.75 on 21 degrees of freedom
AIC: 209.16
Number of Fisher Scoring
iterations: 4
Notice that:
-
from the residual deviance, the model fits well
-
given the significant interaction effects, we can see
that the linear effect of log(doserate)
is different depending on the level of
doseamt
-
its linear effect, or slope, is
0.07178
for doseamt=1,
0.07178+0.16122=0.233
for doseamt=2.5,
and 0.07178+0.19350=0.26528
for doseamt=5.
The slopes increase with doseamt.
-
Q: if we use linear-quadratic
coding for doseamt,
we would expect that the linear-by-linear and linear-by-quadratic
interactions between log(doserate)
and doseamt would
be significant. How can you interpret the significance of the two
interactions? Compare it with the previous interpretation for interactions
coded based on
dummy variables. Which interpretation is easier to understand for you?
-
the combination of a high
doseamt and a high
doserate has a greater
combined effect than the main effect estimates would suggest.
-
exercise: fit a binomial GLM with
logit link to the data and compare its result with this Poisson fitted model
Suppose that we are not sure whether it is reasonable to set
the coefficient of log(nx)
as one. We can allow a parameter for the predictor as follows:
log(mx)=g×log(nx)+Xbºhx'
Û
log(mx/nxg)=hx=Xb.
This is a Poisson GLM with log(nx)
as an ordinary predictor:
> pmod <- glm(ca ~ log(cells)+log(doserate)*dosef, family=poisson,dicentric)
>
summary(pmod)
Call:
glm(formula = ca ~
log(cells) + log(doserate) * dosef, family = poisson, data = dicentric)
Deviance Residuals:
Min 1Q
Median 3Q Max
-1.49901 -0.62229
-0.05021 0.76919 1.59525
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept)
-2.76534 0.38116 -7.255 4.02e-13 ***
log(cells)
1.00252 0.05137 19.517 < 2e-16 ***
log(doserate)
0.07200 0.03547 2.030 0.042403 *
dosef2.5
1.62984 0.10273 15.866 < 2e-16 ***
dosef5
2.76673 0.12287 22.517 < 2e-16 ***
log(doserate):dosef2.5
0.16111 0.04837 3.331 0.000866 ***
log(doserate):dosef5
0.19316 0.04299 4.493 7.03e-06 ***
---
Signif. codes: 0 '***'
0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter
for poisson family taken to be 1)
Null deviance:
916.127 on 26 degrees of freedom
Residual deviance:
21.748 on 20 degrees of freedom
AIC: 211.15
Number of Fisher Scoring
iterations: 4
Notice that:
-
the estimated g,
1.00252, is very
close to one, which supports the use of the previous Poisson model
-
not surprisingly, the estimated
b are only slightly
different from the previous Poisson fitted model