Overdispersion (Reading: Faraway (2006, 1st ed.), section 2.11)
Manly (1978) reported a data from an experiment where boxes of trout eggs were buried at five different stream locations and retrieved at four different times. The times are specified by the number of weeks after the original placement. The number of surviving eggs was recorded. Let us first read the data into R and construct a tabulation of the data:
> troutegg <- read.table("troutegg.txt")
> ftable(xtabs(cbind(survive,total) ~ location+period, troutegg))
survive total
location period
1 4 89 94
7 94 98
8 77 86
11 141 155
2 4 106 108
7 91 106
8 87 96
11 104 122
3 4 119 123
7 100 130
8 88 119
11 91 125
4 4 104 104
7 80 97
8 67 99
11 111 132
5 4 49 93
7 11 113
8 18 88
11 0 138
Notice that
in one case, all the eggs survive, while in another, none of the eggs survive
therefore, we would expect that within the range of the predictors in the dataset, the true px can run from 0 to 1, which indicates:
a Normal linear model with yx/nx as the response should not be a good approximation to the binomial model
the choice of link functions could be an important issue for this case
We can obtain the survival proportion in each cell by:
> xtabs((survive/total) ~ location+period, troutegg)
period
location 4 7 8 11
1 0.94680851 0.95918367 0.89534884 0.90967742
2 0.98148148 0.85849057 0.90625000 0.85245902
3 0.96747967 0.76923077 0.73949580 0.72800000
4 1.00000000 0.82474227 0.67676768 0.84090909
5 0.52688172 0.09734513 0.20454545 0.00000000
We can find some rough conclusions from the table:
no matter what length of retrieving time is chosen, location 5 has a survival probability much lower than other locations
the survival probabilities in the other 4 locations are more consistent
it seems that the survival probabilityhas the tendency of decreasing over retrieving time
it is not clear whether there exists some strong interactions between location and period
Let us fit a binomial GLM with only main effects to the data. We now treat location and period as nominal variables although period can be regarded as a quantitative variable:
> troutegg$location <- as.factor(troutegg$location)
> troutegg$period <- as.factor(troutegg$period)
> bmod <- glm(cbind(survive,total-survive) ~ location + period, family=binomial,troutegg)
> summary(bmod)
Call:
glm(formula = cbind(survive, total - survive) ~ location + period, family = binomial, data = troutegg)
Deviance Residuals:
Min 1Q Median 3Q Max
-4.83046 -0.36497 -0.03034 0.61907 3.24340
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 4.6358 0.2813 16.479 < 2e-16 ***
location2 -0.4168 0.2461 -1.694 0.0903 .
location3 -1.2421 0.2194 -5.660 1.51e-08 ***
location4 -0.9509 0.2288 -4.157 3.23e-05 ***
location5 -4.6138 0.2502 -18.439 < 2e-16 ***
period7 -2.1702 0.2384 -9.103 < 2e-16 ***
period8 -2.3256 0.2429 -9.573 < 2e-16 ***
period11 -2.4500 0.2341 -10.466 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 1021.469 on 19 degrees of freedom
Residual deviance: 64.495 on 12 degrees of freedom
AIC: 157.03
Number of Fisher Scoring iterations: 5
Notice that the deviance of 64.495 on 12 degrees of freedom shows that this model does not fit. Some possible causes of the large deviance are:
sparse data
outlier
wrong Xb structure (the current model only contains main effects)
overdispersion
Before we conclude that there is an overdispersion, we need to eliminate other potential explanations.
With about 100 eggs in each box, we have no problem with sparseness.
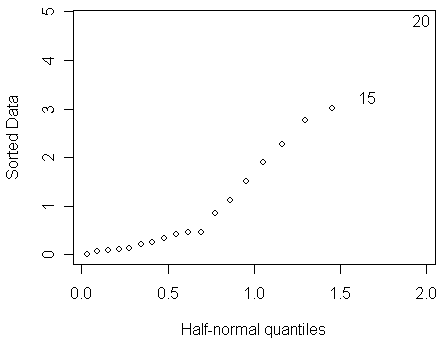
To check for outliers, a half-normal plot of the residuals is a good way. Here is a function for drawing half-normal plot written by Prof. Faraway:
> halfnorm(residuals(bmod))
From the half-normal plot, we find that:
no single outlier is apparent
perhaps one can discern a larger number of residuals which seem to follow a more dispersed distribution than the rest.
For the Xb structure, we can add the 3´4=12 interaction effects to the current main-effect model. After doing this, the new model is a saturated model so that its deviance is zero. In other words, the deviance decreases by 64.495 for 12 degrees of freedom, which of course is statistically significant in terms of the difference-in-deviance test. However, let us draw an interaction plot by using as the response the empirical logits, which can be regarded as empirical estimates of hx=logit(px) and are defined as:
log[(yx+0.5)/(nx-yx+0.5)].
The 0.5 are added to prevent infinite values when nx=yx.
> elogits <- log((troutegg$survive+0.5)/(troutegg$total-troutegg$survive+0.5))
> with(troutegg,interaction.plot(period,location,elogits))
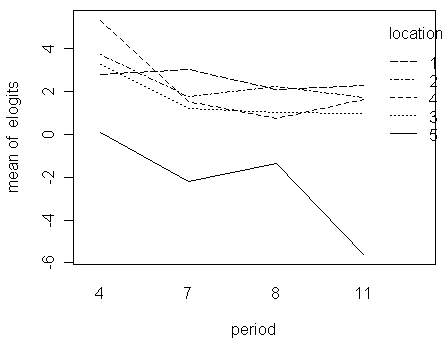
Notice that:
interaction plots are always difficult to interpret conclusively
for this case, we can say the five lines are roughly parallel to each other if the sharp decrease in the end of the location-5 line is ignored; in other words, there is no obvious sign of large interactions
we might conclude that although interactions are statistically significant, they are not physically very significant
so, there is no clear evidence that the Xb structure with only main effects is inadequate
Having eliminated these more obvious causes of large deviance, we may put the blame on overdispersion. Possible reasons for the overdispersion include:
heterogeneous trout eggs
variation in the experimental procedures
unknown variables affecting survival probability
...
The simplest approach for modeling overdispersion is to introduce an additional dispersion parameter s2. We can estimate the dispersion parameter as:
> (sigma2 <- sum(residuals(bmod,type="pearson")^2)/12)
[1] 5.330322
We see that this is substantially larger than one as it would be in the standard binomial GLM.
We can now make F-test (c.f., chi-square test for standard binomial GLM) on the predictors using:
> drop1(bmod,scale=sigma2,test="F")
Single term deletions
Model:
cbind(survive, total - survive) ~ location + period
scale: 5.330322
Df Deviance AIC F value Pr(F)
<none> 64.50 157.03
location 4 913.56 308.32 39.494 8.142e-07 ***
period 3 228.57 181.81 10.176 0.001288 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Warning message:
F test assumes 'quasibinomial' family in: drop1.glm(bmod, scale = sigma2, test = "F")
Notice that:
both predictors are significant (note: we can see from the interaction plots that some main effects can be removed to further simplify the model)
in the command, it is necessary to specify the scale argument using the estimated value of s2
if this scale argument is omitted, the deviance, rather than Pearson X2, will be used in the estimation of s2
this makes very little difference in this particular dataset
but, in some cases, using the deviance instead of Pearson X2 to estimate s2 gives inconsistent results
the warning message reminds us that the use of s2 results in a model that is no longer a standard binomial GLM
no goodness-of-fit test is possible because we now have a free dispersion parameter s2
To test the significance of individual parameters, we can use the estimated s2 to scale up the standard errors of parameter estimates:
> summary(bmod,dispersion=sigma2)
Call:
glm(formula = cbind(survive, total - survive) ~ location + period, family = binomial, data = troutegg)
Deviance Residuals:
Min 1Q Median 3Q Max
-4.83046 -0.36497 -0.03034 0.61907 3.24340
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 4.6358 0.6495 7.138 9.49e-13 ***
location2 -0.4168 0.5682 -0.734 0.4632
location3 -1.2421 0.5066 -2.452 0.0142 *
location4 -0.9509 0.5281 -1.800 0.0718 .
location5 -4.6138 0.5777 -7.987 1.39e-15 ***
period7 -2.1702 0.5504 -3.943 8.05e-05 ***
period8 -2.3256 0.5609 -4.146 3.38e-05 ***
period11 -2.4500 0.5405 -4.533 5.82e-06 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 5.330322)
Null deviance: 1021.469 on 19 degrees of freedom
Residual deviance: 64.495 on 12 degrees of freedom
AIC: 157.03
Number of Fisher Scoring iterations: 5
(exercise:
compare the results with the output of the standard binomial GLM
find their differences
explain how the differences affect the conclusions.)
(Q: should we still use Normal(0,1) to calculate the p-values? is t-distribution a better choice?)